...
The simulation seems to be running slowly because there are too many neighbours on the program, it seems that for the central atoms the number of neighbors is more than 2000. This may be caused because of the electrostatic potential cutoff. (Not so slow)
AWSEM-3SPN.2 doesn't run with the new version (it almost runs, but atoms from AWSEM dissapear, with the same configuration that worked on the old version) (Using the old version)
The premerge script also needs some modifications if I want to simulate the RNA in A-form. Currently it is impossible to simulate RNA with the A-form forcefield, but the 3SPN.2C seems to keep the RNA structure. (Using the B-form, seems to keep the shape if the RNA template used is the native conformation)
It is impossible to restart the simulations, since the read_restart doesn't work with 3SPN. (A possible fix may be on the new lammps version with read_restart remap) (Still imposible)
To Do
Simulation of the complex with a SMF of 0.1 (may be more rigid) (Running)
Use fragment memory force for the parts of the protein that are flexible. (Running simulations with dual memory, with a MF of 0.05. Should be equivalent to a simulation of SMF 0.1 but with two 0.05 wells around the region where the conformation is diferent. Running with and without DNA)
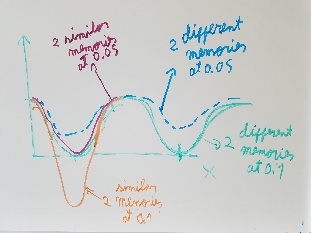
Run multiple simulations and analyze the PC, see that there are many transitions
Run simulations without DNA (Maybe have another fragment of the structure without RNA)
Check which crystal is closer to the DNA and do bias simulations?