...
Use fragment memory force for the parts of the protein that are flexible. (Running simulations with dual memory, with a MF of 0.05. Should be equivalent to a simulation of SMF 0.1 but with two 0.05 wells around the region where the conformation is diferent. Running with and without DNA)
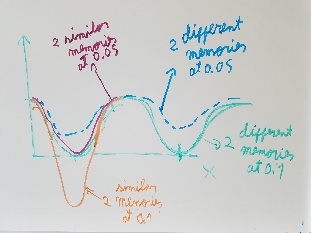
Run multiple simulations and analyze the PC, see that there are many transitions
Run simulations without DNA (Maybe have another fragment of the structure without RNA)
Check which crystal is closer to the DNA and do bias simulations?