Crispr-Cas9 (Apo form)
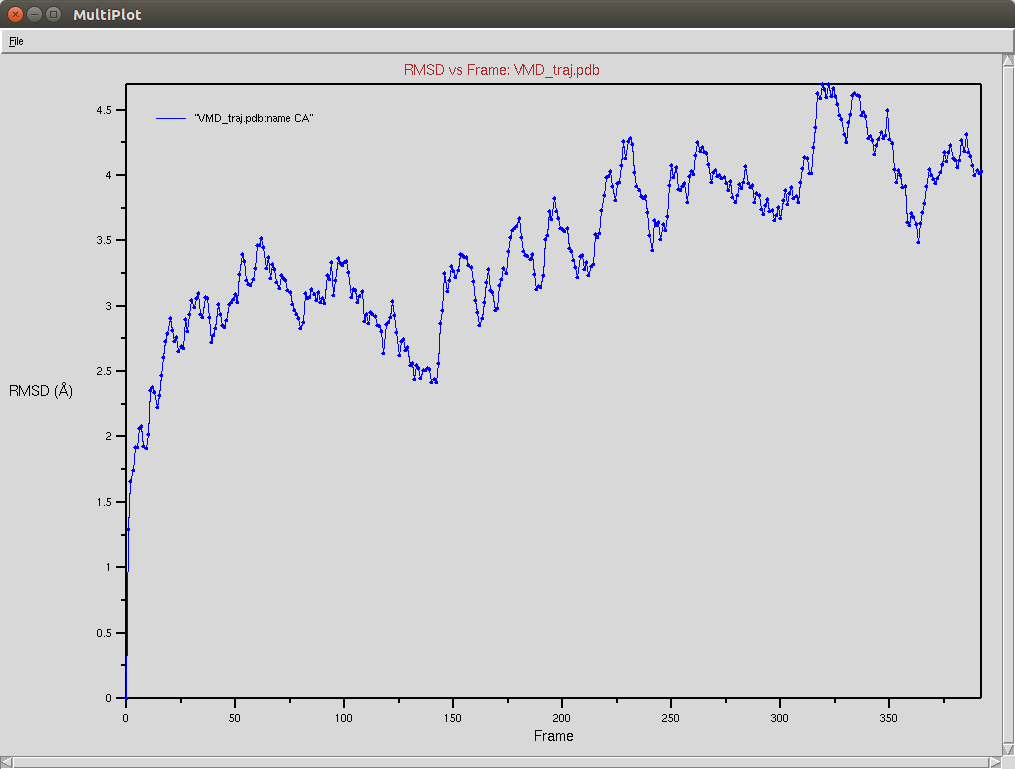
- The framgent memory force in this simulation was too big (0.2), I'm repeating the simulation with a smaller force (0.01). I expect that the energy of the local interactions (RAMA+HELIX+F.M.) should contribute around 2/3 of the energy of the non-local interactions (DSSP,PAP,WATER)
Protein RMSD
In order to calculate a reasonable SMF Nick and me analyzed some simulations of T4 Lysozyme, where we can see the change of Energy with the Q-value. On this simulations it is possible to find Lysozymes with Qvalues between 0.4 and 0.98. We can use this values to estimate an energy change for any protein.

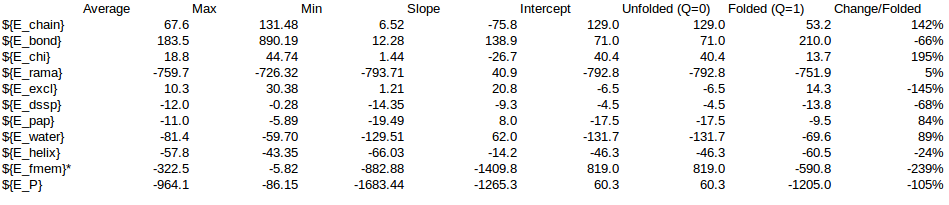
/home/cab22/Documents/Lysozyme/1lw9_Unfolding_Qvalue/Energy_Comparison.ods
As a rule of thumb we expect that the energy coming from the local interactions should be around 1/3 of the local interactions. We can use the expectations to approximate the energy change in the Unfolding of CRISPR.
| Energy | Energy change | between Q=1 and Q=0 | |||
| SMF=0.01 | SMF=0.2 | Change_expected | SMF=0.01 | SMF=0.2 | |
| ${Step} | 1000 | 1000 | |||
| ${E_chain} | 1103.63 | 1066.13 | 142.39% | 1571.49 | 1518.09 |
| ${E_bond} | 3719.19 | 3591.47 | -66.16% | -2460.76 | -2376.26 |
| ${E_chi} | 311.13 | 270.81 | 194.72% | 605.81 | 527.30 |
| ${E_rama} | -5324.09 | -5215.35 | 5.44% | -289.41 | -283.50 |
| ${E_excl} | 70.33 | 111.11 | -145.26% | -102.16 | -161.40 |
| ${E_dssp} | -76.22 | -102.85 | -67.69% | 51.59 | 69.62 |
| ${E_pap} | -78.58 | -93.15 | 83.73% | -65.79 | -78.00 |
| ${E_water} | -752.31 | -524.77 | 89.06% | -670.04 | -467.38 |
| ${E_helix} | -229.17 | -283.44 | -23.52% | 53.90 | 66.67 |
| ${E_fmem} | -54.26 | -5632.65 | 100.00% | -54.26 | -5632.65 |
| ${E_P} | -1310.35 | -6812.70 | -105.00% | 1375.92 | 7153.62 |
| Local | -5607.52 | -11131.44 | -289.77 | -5849.49 | |
| NonLocal | -907.11 | -720.78 | -684.24 | -475.77 | |
| Others | 5204.27 | 5039.52 | -385.62 | -492.27 | |
| Local+Non-local | -974.0133132415 | -6325.2556957897 | |||
| Local% | 29.75% | 92.48% | |||
| NonLocal% | 70.25% | 7.52% |
Crispr-Cas9 + DNA
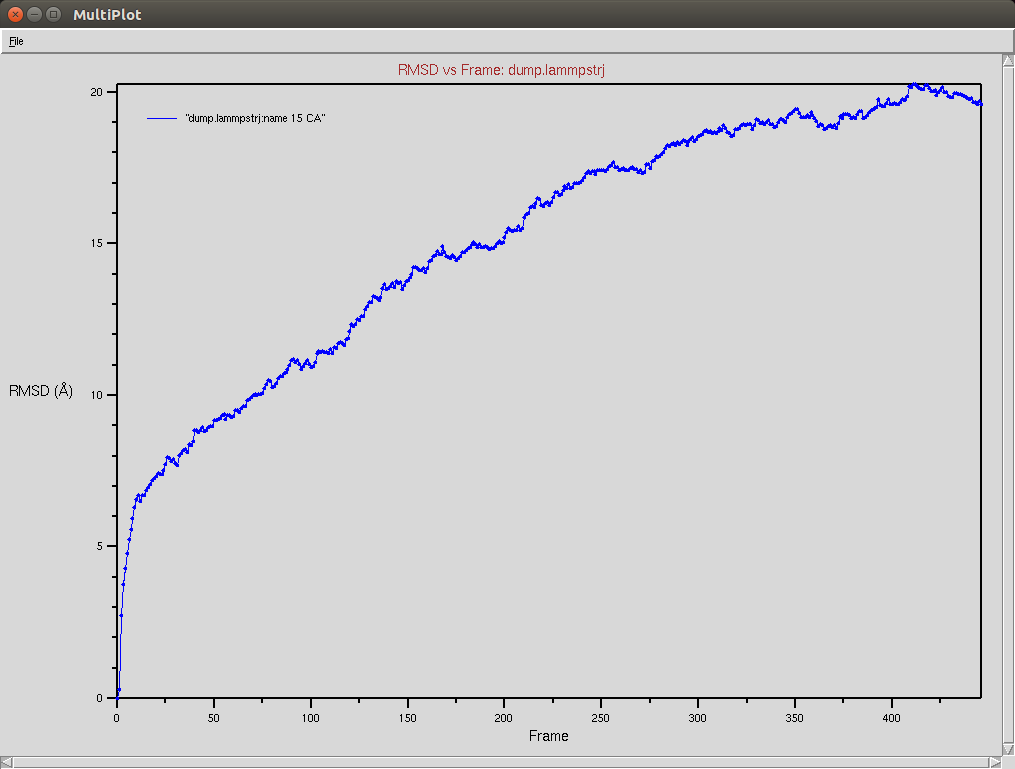
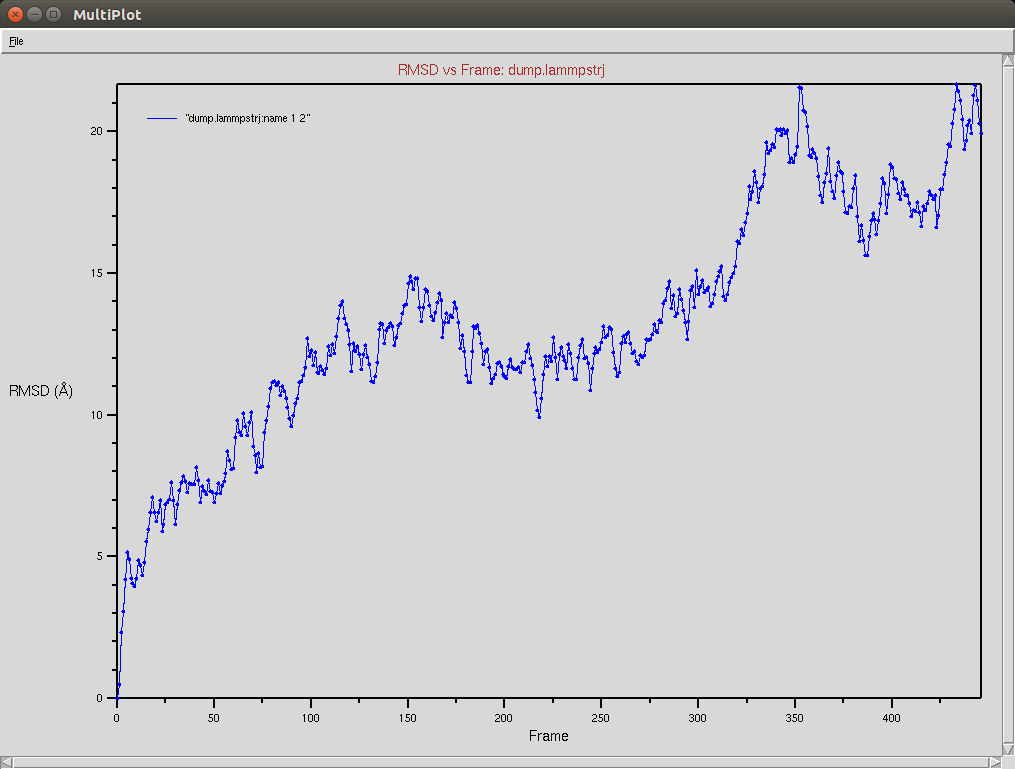
Due to a mistake I didn't update the neighbor list, so it is possible to see an overlap between two domains (grey and red). The single memory force was turned off in this simulation.
Protein RMSD
Nucleic RMSD
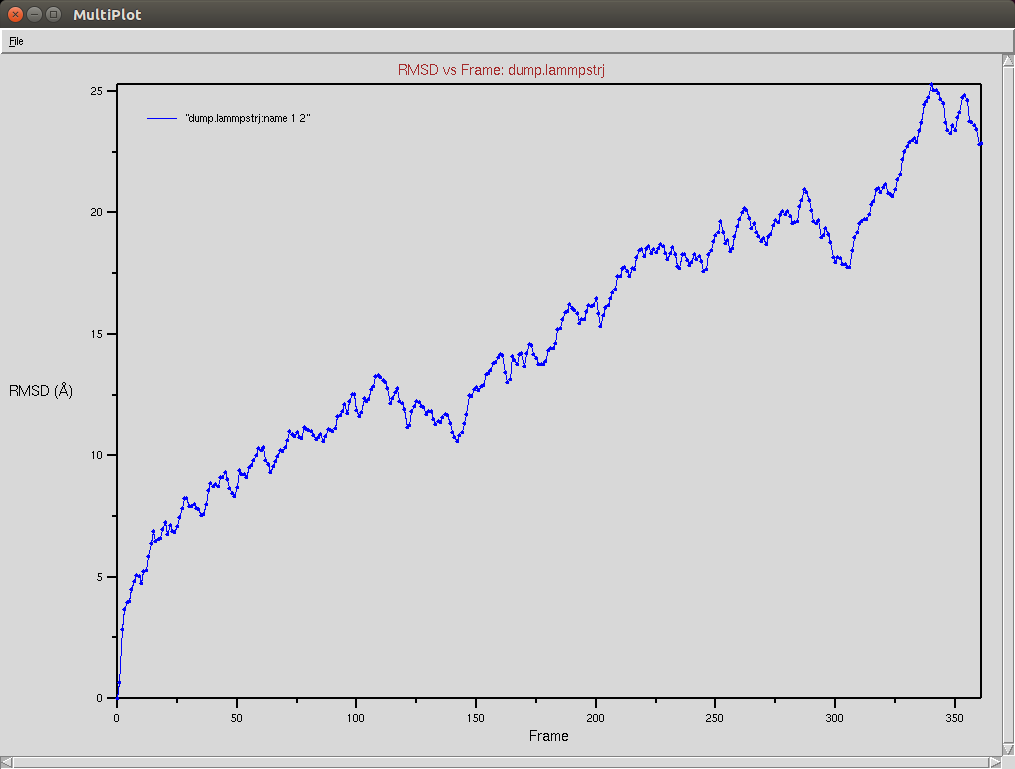
Crispr-Cas9 + RNA
Due to a mistake I didn't update the neighbor list, so it is possible to see an overlap between two domains (grey and red). The single memory force was turned off in this simulation.
Protein RMSD
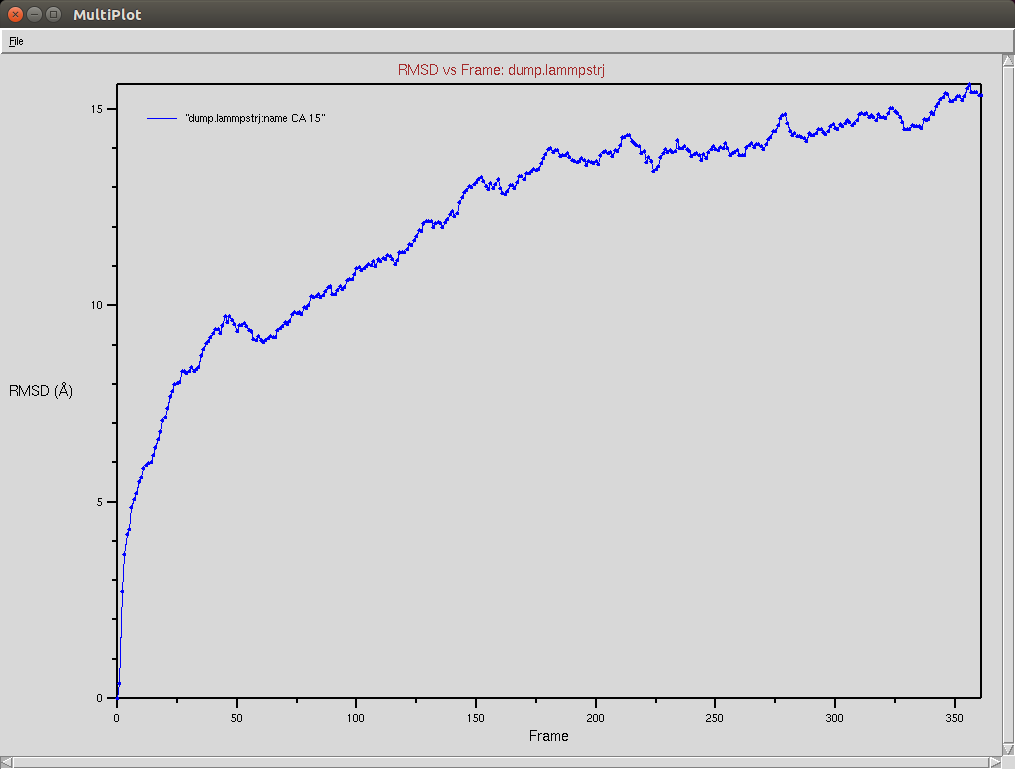
Nucleic RMSD
Some technical constraints
It is still difficult to mix two nucleic chain together (like DNA and RNA) because merge.py only accepts two files. (Working on this)
The simulation seems to be running slowly because there are too many neighbours on the program, it seems that for the central atoms the number of neighbors is more than 2000. This may be caused because of the electrostatic potential cutoff.
AWSEM-3SPN.2 doesnt run with the new version (it almost runs, but atoms from AWSEM dissapear, with the same configuration that worked on the old version)
The premerge script also needs some modifications if I want to simulate the RNA in A-form. Currently it is impossible to simulate RNA with the A-form forcefield, but the 3SPN.2C seems to keep the RNA structure.
It is impossible to restart the simulations, since the read_restart doesn't work with 3SPN. (A possible fix may be on the new lammps version with read_restart remap)