1. Introduction
PhyloNet is a tool designed mainly for analyzing, reconstructing, and evaluating reticulate (or non-treelike) evolutionary relationships, generally known as phylogenetic networks. Various methods that we have developed make use of techniques and tools from the domain of phylogenetic networks, and hence the PhyloNet package includes several tools for phylogenetic network analysis. PhyloNet is released under the GNU General Public License. For the full license, see the file GPL.txt included with this distribution.
PhyloNet is designed, implemented, and maintained by Rice's BioInformatics Group, which is lead by Professor Luay Nakhleh (nakhleh@cs.rice.edu). For more details related to this group please visit http://bioinfo.cs.rice.edu.
This tutorial is based on the book chapter: Practical Aspects of Phylogenetic Network Analysis Using PhyloNet. When you try the examples, we suggest you refer to the corresponding section of the book chapter.
2. Installation
System Requirements
In order to run the PhyloNet toolkit, you must have Java 1.8.0 or later installed on your system. All references to the java command assume that Java 1.7 is being used.
- To check your Java version, type "java -version" on your command line.
- To download Java 1.8, please go to website http://www.java.com/en/download/.
To link to the new downloaded Java 1.8, for mac, try these two commands from command line:
sudo rm /usr/bin/java sudo ln -s /Library/Internet\ Plug-Ins/JavaAppletPlugin.plugin/Contents/Home/bin/java /usr/bin
Downloading phylonet.jar
Acquire the current release of PhyloNet by downloading the most recent version of the PhyloNet JAR file. You will have a file named PhyloNet_X.Y.Z.jar, where X is the major version number and Y and Z are the minor version numbers.
Installing the file
Place the jar file in the desired installation directory. The remainder of this document assumes that it is located in directory $PHYLONET_DIRECTORY. Installation is now complete. In order to run PhyloNet, you must execute the file PhyloNet_X.Y.Z.jar, as described in the next section.
3. Basic Usage
The PhyloNet tool is executed by typing the following command into your console:
java -jar $PHYLONET_DIRECTORY/PhyloNet_X.Y.Z.jar script.nex
Where $PHYLONET_DIRECTORY is the directory of jar file PhyloNet_X.Y.Z.jar, and script.nex is the NEXUS file containing the commands to be executed.
Scoring a candidate species phylogeny
- Parsimonious:
- Scoring a species tree (ILS): DeepCoalCount_tree
- Scoring a species tree/network (ILS+introgression): DeepCoalCount_network
- Probabilistic:
- Scoring a species tree/network (ILS+introgression): CalGTProb
Inferring a species phylogeny
- Maximum parsimony:
- Inferring a species tree (ILS): Infer_ST_MDC, Infer_ST_MDC_UR
- Inferring a species tree/network (ILS+introgression): InferNetwork_MP
- Maximum likelihood:
- Inferring a species tree/network (ILS+introgression): InferNetwork_ML
- inferring a species tree/network (ILS+introgression) with cross-validation: InferNetwork_ML_CV
- Maximum pseudo-likelihood:
- Inferring a species tree/network (ILS+introgression): InferNetwork_MPL
- Bayesian:
- Inferring a species tree/network (ILS+introgression): MCMC_GT
- Inferring a species tree/network (ILS+introgression) from multilocus data: MCMC_SEQ
- Inferring a species tree/network (ILS+introgression) from bi-allelic marker data: MCMC_BiMarkers
- Larger data sets:
- Inferring a species tree/network (ILS+introgression) by divide-and-conquer: NetMerger
- Inferring a species tree/network (ILS+introgression) while fixing the start tree topology: InferNetwork_MPL (-fs) and InferNetwork_MP (-fs)
Comparing and Summarizing
- Summarizing networks: SummarizeNetworks
For other tools in PhyloNet, please see here for a full list.
4. Illustrating the Various Inference Methods in PhyloNet
Here we provide all the input NEXUS files in section 4 of the book chapter. The figure below is the true network we would like to infer.
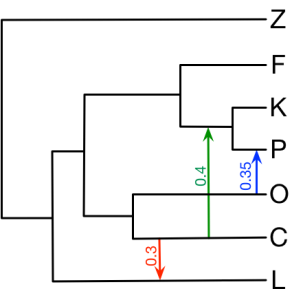
4.1 Minimize deep coalescent Inference
This section corresponds to section 4.1 of the book chapter.
4.1.1 MDC Inference using true gene tree topologies
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_MP_pl8_0_true.nex | 0 |
| InferNetwork_MP_pl8_1_true.nex | 1 |
| InferNetwork_MP_pl8_2_true.nex | 2 |
| InferNetwork_MP_pl8_3_true.nex | 3 |
| InferNetwork_MP_pl8_4_true.nex | 4 |
Corresponding results: Fig 4 in the book chapter.
4.1.2 MDC Inference using gene tree topologies estimated by IQTREE
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_MP_pl8_0_false.nex | 0 |
| InferNetwork_MP_pl8_1_false.nex | 1 |
| InferNetwork_MP_pl8_2_false.nex | 2 |
| InferNetwork_MP_pl8_3_false.nex | 3 |
| InferNetwork_MP_pl8_4_false.nex | 4 |
Corresponding results: Fig 5 in the book chapter.
4.2 Maximum Likelihood Inference
This section corresponds to section 4.2 of the book chapter.
4.2.1 ML Inference using true gene tree topologies
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_ML_pl8_0_true.nex | 0 |
| InferNetwork_ML_pl8_1_true.nex | 1 |
| InferNetwork_ML_pl8_2_true.nex | 2 |
| InferNetwork_ML_pl8_3_true.nex | 3 |
| InferNetwork_ML_pl8_4_true.nex | 4 |
Corresponding results: Fig 6 in the book chapter.
4.2.2 ML Inference using gene tree topologies estimated by IQTREE
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_ML_pl8_0_false.nex | 0 |
| InferNetwork_ML_pl8_1_false.nex | 1 |
| InferNetwork_ML_pl8_2_false.nex | 2 |
| InferNetwork_ML_pl8_3_false.nex | 3 |
| InferNetwork_ML_pl8_4_false.nex | 4 |
Corresponding results: Fig 6 in the book chapter.
4.2.3 ML Inference using true gene tree topologies and branch lengths
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_ML_bl_pl8_0_true.nex | 0 |
| InferNetwork_ML_bl_pl8_1_true.nex | 1 |
| InferNetwork_ML_bl_pl8_2_true.nex | 2 |
| InferNetwork_ML_bl_pl8_3_true.nex | 3 |
| InferNetwork_ML_bl_pl8_4_true.nex | 4 |
Corresponding results: Fig 7 in the book chapter.
4.2.4 ML Inference using gene tree topologies and branch lengths estimated by IQTREE
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_ML_bl_pl8_0_false.nex | 0 |
| InferNetwork_ML_bl_pl8_1_false.nex | 1 |
| InferNetwork_ML_bl_pl8_2_false.nex | 2 |
| InferNetwork_ML_bl_pl8_3_false.nex | 3 |
| InferNetwork_ML_bl_pl8_4_false.nex | 4 |
Corresponding results: Fig 9 in the book chapter.
4.3 Maximum Pseudo-likelihood Inference
This section corresponds to section 4.3 of the book chapter.
4.3.1 MPL Inference using true gene tree topologies
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_MPL_pl8_0_true.nex | 0 |
| InferNetwork_MPL_pl8_1_true.nex | 1 |
| InferNetwork_MPL_pl8_2_true.nex | 2 |
| InferNetwork_MPL_pl8_3_true.nex | 3 |
| InferNetwork_MPL_pl8_4_true.nex | 4 |
Corresponding results: Fig 10 in the book chapter.
4.3.2 MPL Inference using gene tree topologies estimated by IQTREE
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_MPL_pl8_0_false.nex | 0 |
| InferNetwork_MPL_pl8_1_false.nex | 1 |
| InferNetwork_MPL_pl8_2_false.nex | 2 |
| InferNetwork_MPL_pl8_3_false.nex | 3 |
| InferNetwork_MPL_pl8_4_false.nex | 4 |
Corresponding results: Fig 11 in the book chapter.
4.3.3 MPL Inference using bi-allelic marker data
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| mle_bi_0pseudo.nexus | 0 |
| mle_bi_1pseudo.nexus | 1 |
| mle_bi_2pseudo.nexus | 2 |
| mle_bi_3pseudo.nexus | 3 |
| mle_bi_4pseudo.nexus | 4 |
Corresponding results: Fig 12 in the book chapter.
4.4 Bayesian Inference
This section corresponds to section 4.4 of the book chapter.
4.4.1 MCMC_SEQ: Bayesian inference on the sequence alignment data
To use MCMC_SEQ, you need to download an additional package beagle to calculate Felsenstein Likelihood. Please follow "Installing from source".
Add this package to your java library, and load this library before you run PhyloNet.
One input example: mcmc_seq.nex
All input NEXUS files: download
Corresponding results: Fig 13 in the book chapter.
4.4.2 MCMC_GT: Bayesian inference on gene tree topologies
4.4.2.1 MCMC_GT sampling using true gene tree topologies
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| MCMC_GT_pl8_0_true.nex | 0 |
| MCMC_GT_pl8_1_true.nex | 1 |
| MCMC_GT_pl8_2_true.nex | 2 |
| MCMC_GT_pl8_3_true.nex | 3 |
| MCMC_GT_pl8_4_true.nex | 4 |
This inference ran out of 192 CPU hours.
4.4.2.2 MCMC_GT sampling using gene tree topologies estimated by IQTREE
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| MCMC_GT_pl8_0_false.nex | 0 |
| MCMC_GT_pl8_1_false.nex | 1 |
| MCMC_GT_pl8_2_false.nex | 2 |
| MCMC_GT_pl8_3_false.nex | 3 |
| MCMC_GT_pl8_4_false.nex | 4 |
This inference ran out of 192 CPU hours.
4.4.3 MCMC_BiMarkers: Bayesian inference on the bi-allelic markers
This method uses an additional package jeigen. You need to follow the instructions to install it, and add this package to your java library.
Input NEXUS file: mcmc_bimarker.nexus
This inference ran out of 192 CPU hours.
4.5 Analyzing Larger Data Sets
This section corresponds to section 5 in the book chapter.
4.5.1 Tree-based Augmentation
The -fs command in MP and MPL is to fix the start tree topology.
The following two examples are to infer a network using gene trees estimated by IQTREE and fixing the start species tree inferred by ASTRAL.
Input NEXUS file for MP: InferNetwork_MP_pl8_3_false_fs.nex
Input NEXUS file for MPL: InferNetwork_MPL_pl8_3_false_fs.nex
4.5.2 Divide-and-conquer
The data set contains the MCMC_SEQ outputs of 680 trinets. You need to download the data and change the path ".../DivideAndConquer/" in netmerger.nex.
Data: https://drive.google.com/file/d/1mJfqD0bQOOoBFZTlaQklHLqvJX5QPlVg/view?usp=sharing
Input NEXUS file: netmerger.nex
4.6 Analyzing Polyploids
This section corresponds to section 7 in the book chapter.
4.6.1 MDC Inference with unknown hybrid species
| Input NEXUS file | Maximum number of reticulations |
|---|---|
| InferNetwork_MP_0.nex | 0 |
| InferNetwork_MP_1.nex | 1 |
| InferNetwork_MP_2.nex | 2 |
| InferNetwork_MP_3.nex | 3 |
Corresponding results: Fig 17 in the book chapter.
4.6.2 MDC Inference with known hybrid species
| Input NEXUS file | Maximum number of reticulations | Specified hybrid species |
|---|---|---|
| InferNetwork_MP_1.nex | 1 | LPS168 |
| InferNetwork_MP_2.nex | 2 | LPS168 |
| InferNetwork_MP_2_2.nex | 2 | LPS168, LPS189 |
| InferNetwork_MP_3.nex | 3 | LPS168 |
| InferNetwork_MP_3_2.nex | 3 | LPS168, LPS189 |
Corresponding results: Fig 18 in the book chapter.
5. Visualizing a Phylogenetic Network
Phylogenetic network in Rich Newick string can be visualized in Dendroscope or icytree. The former needs downloading, and the latter is online. However, Dendroscope cannot recognize inheritance probabilities (branch lengths are fine), and icytree sometimes can and sometimes cannot. You need to remove those probabilities manually from the Rich Newick string, or use option "-di" so that PhyloNet returns the network that Dendroscope takes directly. For example, the following network is what PhyloNet returns when the input NEXUS file InferNetwork_ML_pl8_1_true.nex is used:
((F:12.842,((L:5.576504712905155,((O:1.2260000000000002,P:1.2260000000000002)I6:0.8559999999999997,K:2.082)I2:3.4945047129051554)I0:0.08027116443257487)I8#H1:7.18522412266227::0.4334172270375128)I1:19.128,(I8#H1:1.81722412266227::0.5665827729624873,C:7.474)I4:24.496000000000002)I7;
After removing "::" and the number after it, we have the network below, which can be visualized in both tools.
((F:12.842,((L:5.576504712905155,((O:1.2260000000000002,P:1.2260000000000002)I6:0.8559999999999997,K:2.082)I2:3.4945047129051554)I0:0.08027116443257487)I8#H1:7.18522412266227)I1:19.128,(I8#H1:1.81722412266227,C:7.474)I4:24.496000000000002)I7;
6. References
- . PhyloNet: A Software Package for Analyzing and Reconstructing Reticulate Evolutionary Relationships. BMC Bioinformatics, 9:322, 2008
- C. Than and L. Nakhleh. Species tree inference by minimizing deep coalescences. PLoS Computational Biology, 5(9):e1000501, 2009.
- Y. Yu, T. Warnow, and L. Nakhleh. Algorithms for MDC-based multi-locus phylogeny inference. Proceedings of the 15th Annual International Conference on Research in Computational Molecular Biology (RECOMB), LNBI 6577, 531-545, 2011.
- Y. Yu, T. Warnow, and L. Nakhleh. Algorithms for MDC-based multi-locus phylogeny inference: Beyond rooted binary gene trees on single alleles. Journal of Computational Biology, 18(11):1-18, 2011.
- Y. Yu, J.H. Degnan, and L. Nakhleh. The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genetics, 8(4):e1002660, 2012.
- Y. Yu, R.M. Barnett, and L. Nakhleh. Parsimonious inference of hybridization in the presence of incomplete lineage sorting. Systematic Biology, vol. 62, no. 5, pp. 738-751, 2013.
- Y. Yu and L. Nakhleh. Fast algorithms for reconciliation under hybridization and incomplete lineage sorting. BMC Bioinformatics, vol. 14, no. Suppl 15, p. S6, 2013.
- Y. Yu, J. Dong, K. Liu, and L. Nakhleh, “Probabilistic inference of reticulate evolutionary histories,” Proceedings of the National Academy of Sciences, vol. 111, no. 46, pp. 16448-16453, 2014.
- , “A Maximum Pseudo-likelihood Approach for Phylogenetic Networks”, BMC Genomics, vol. 16, no. Suppl 10, p. S10, 2015.
- , “Reticulate Evolutionary History and Extensive Introgression in Mosquito Species Revealed by Phylogenetic Network Analysis”, Molecular Ecology, vol. 25, pp. 2361-2372, 2016.
- , “Bayesian inference of species phylogenies under the multispecies network coalescent”, PLoS Genetics, vol. 12, no. 5, p. e1006006, 2016.
- D.Wen and L. Nakhleh. Co-estimating reticulate phylogenies and gene trees on sequences from multiple independent loci. Systematic Biology 67.3 (2017): 439-457.
- Z. Cao, X. Liu, HA. Ogilvie, Z. Yan, and L. Nakhleh. Practical aspects of phylogenetic network analysis using phylonet. bioRxiv (2019): 746362.
- Z. Cao, J. Zhu, and L. Nakhleh. Empirical Performance of Tree-Based Inference of Phylogenetic Networks. WABI 2019.